Brar Lab Research
Explore current projects by clicking on icons below
Click here for more on protein degradation and translation

Click here for more on stress pathways
Click here for more on uORFs and transcript isoforms in meiosis
Click here for more on meiotic sORFs
Overview
We aim to unravel the unconventional molecular circuitry that allows precursor cells to be remodeled into gametes.
Regulation of gene expression allows cells to grow, adapt to their environment, and differentiate into new forms. We know the key processes responsible—including transcription, translation, and protein degradation—but have a poor understanding of how they are naturally deployed and integrated to drive cellular change. The Brar lab studies the gene regulatory circuitry that underlies meiotic differentiation, the process that creates gametes. This conserved process involves dramatic and characteristic changes to cell morphology and function that reflect a series of unidirectional transitions in cell state. Meiosis fails frequently, with devastating consequences for fertility and health, but we do not understand the basis of these failures, due in part to gaps in our understanding of normal regulation. My lab has constructed an atlas of gene expression through meiosis, providing a framework to begin to explain its molecular control.
Our studies have also revealed new principles in gene regulation that may broadly apply to natural conditions of cellular change, and that have been missed by model-centric analyses. Canonical models for gene regulation are based on studies of individual genes and rational assumptions, such as a focus on cellular economy. They predate our ability to make genome-scale measurements. Such models tell us, for example, that proteins should be translated from Open Reading Frames (ORFs) that exceed 100 codons and that ORFs should initiate at AUG codons. Cases in which these rules do not fit have largely been treated as exceptions. Our studies of meiosis have shown that both the regions that are expressed and how they are regulated are not well explained by canonical gene regulatory rules. Systems-level studies, complemented with classical approaches, have allowed us to determine that unexpected patterns reflect substantial gaps in our understanding of how gene expression is naturally regulated. We are investigating the basis for these surprising findings, with the goal of discovering new gene regulatory features and explaining how they shape a meiotic cell. Our approach is to use integrated parallel global gene expression measurements and quantitative analytical frameworks, complemented with classical approaches, to gain fresh insight into eukaryotic gene regulatory circuitry.
The rules that govern how cells naturally use their genomic resources over time remain mysterious. Using the approaches described here, we aim to finally define them. We constructed the most comprehensive map of gene expression through a developmental process, including mRNA, translation, and protein measurements for many naturally synchronous stages of meiotic differentiation in budding yeast. We found strong temporal regulation of synthesis and degradation for nearly every gene in the genome, and these patterns were specific enough to enable accurate prediction of gene function. The highly parallel nature of the measurements enabled discovery of gene regulatory strategies that defy canonical models, including the widespread integration of translational and transcriptional control and the synthesis of thousands of non-canonical proteins.
Brar lab studies
Gene expression is the process by which information encoded in the genome is selectively decoded into proteins. Its regulation is necessary for a cell to grow, adapt to its environment, and differentiate into a new form. Regulation of transcription, or the conversion of DNA-encoded information to mRNA, determines the set of genes that are available for expression as proteins and has been the predominant focus of most gene regulation studies to date. Regulation of translation—the process by which the ribosome converts the nucleotide code in mRNA into amino acid chains—determines the timing of protein production and ultimately defines the level at which each protein is made. Protein degradation is the process by which proteins are removed from the cell and is an important facet of gene regulation, but to only a small proportion of gene products are, to date, known to be subject to regulated degradation. Studies of gene expression, in general, have largely focused on isolated analyses of individual genes or processes. These studies have led to a common and rational set of assumptions about how eukaryotic gene expression is generally regulated, including about the specific features that define a gene and the linear nature of gene expression “steps”. Although, built on these foundational assumptions, we now have a strong understanding of the mechanics of eukaryotic gene expression regulation—including how transcription, translation, and protein degradation work—we have a very poor understanding of how they are naturally regulated and interconnected, particularly during conditions of cellular change.
The development of techniques for measuring gene expression globally has greatly expanded our understanding of gene regulatory mechanisms in depth and scale. These methods finally allow scientists the opportunity to empirically revisit the canonical rules for gene expression in a systematic way and in diverse contexts. We can now quantify key intermediates and transitions in the canonical pathway of gene expression—from DNA to mRNA to protein—genome-wide. Our understanding of translation, in particular, has benefited immensely from the development of a tool—ribosome profiling—that enables its genome-wide measurement in vivo. Ribosome profiling, the deep sequencing of ribosome protected mRNA fragments, monitors protein synthesis with scale, speed, and accuracy rivaling approaches for mRNA measurement (Ingolia et al., Science, 2009; Ingolia et al., Nature Protocols, 2012; Brar and Weissman Nature Reviews Mol Cell Bio, 2015; Brar Cell 2016). We apply this method routinely, including to monitor translation in parallel with mRNA and protein levels at many time points. We have used such studies to generate a rich atlas of meiotic events and gene expression and the first high-resolution map of protein synthesis through a developmental program (Brar et al. Science, 2012; Cheng and Otto et al. Cell, 2018; Eisenberg et al. Cell Systems 2020).

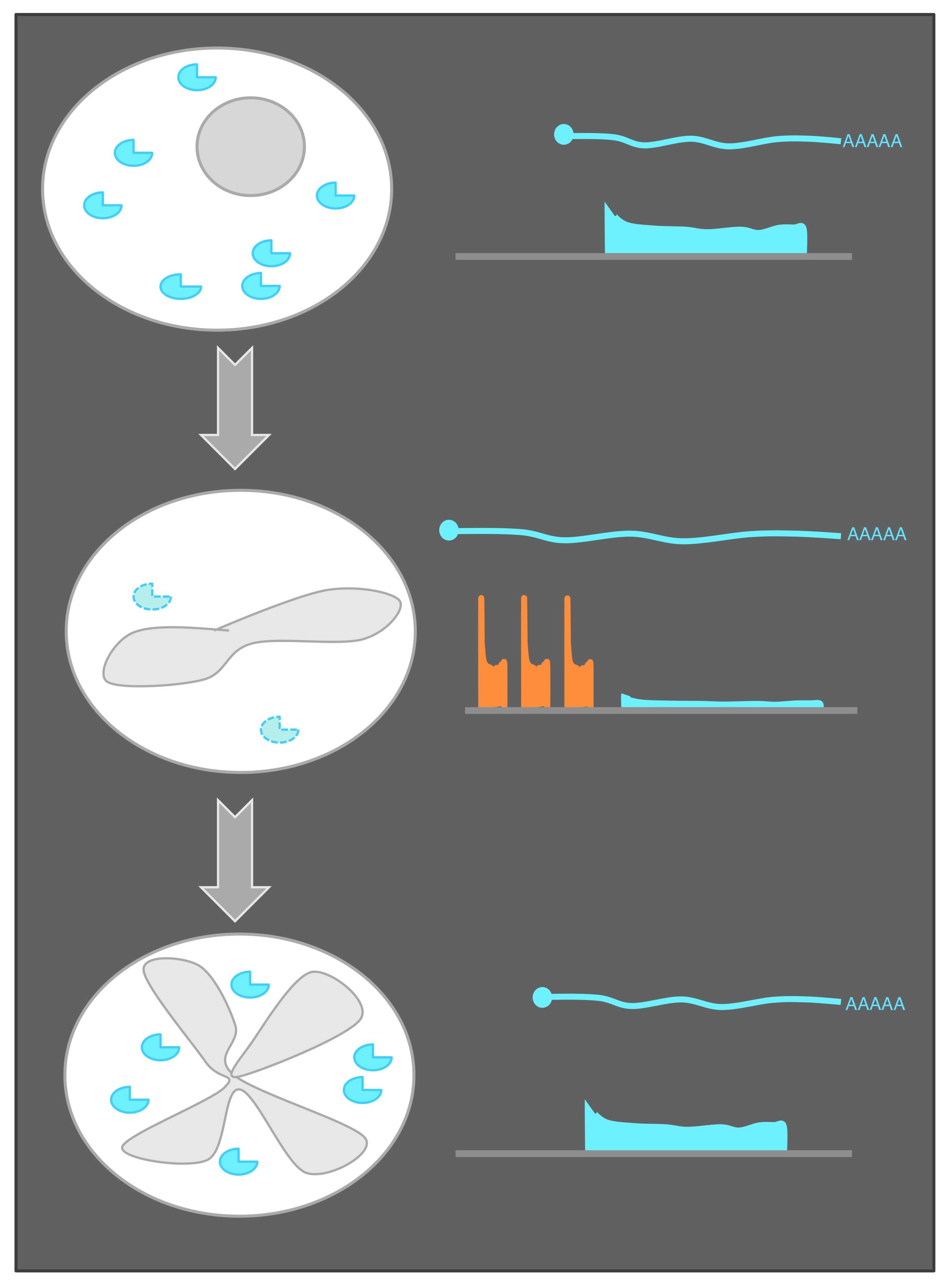
What is ribosome profiling? Nuclease digestion of translating ribosomes protects only mRNA regions (footprints) being actively decoded by the ribosome (Steitz, Nature, 1969; Ingolia et al., Science, 2009). These footprints can be collected in vivo from cells and sequenced to define coding regions and quantify new protein synthesis. Shown is a comparison of the method with mRNA sequencing, which defines the positions of full transcripts, but cannot specifically identify coding regions.
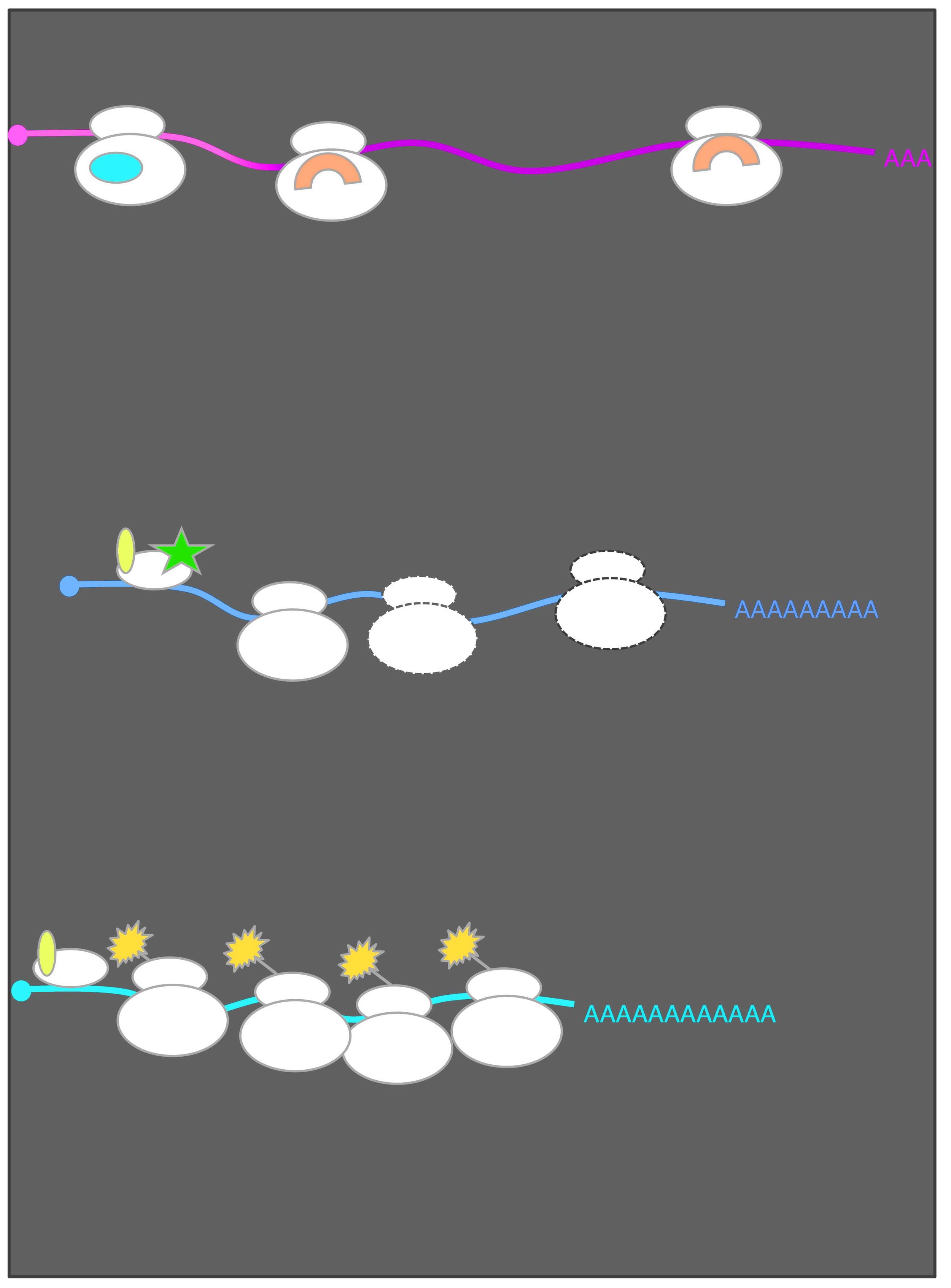
Employing global measurements, including ribosome profiling, in parallel is a powerful approach that my lab has leveraged to paint a more complete picture of the gene regulatory circuitry underlying natural contexts of cellular change (Otto et al. Current Genetics, 2018). In this manner, and with careful experimental design and quantitative analyses in several eukaryotic contexts, our work has provided new and surprising insight into the natural eukaryotic regulation of gene expression. Our studies have revealed an unprecedented view of the molecular events underlying diverse aspects of meiotic biology and uncovered numerous new and dramatic instances of dynamic translational and transcriptional regulation through meiosis. These efforts have also yielded several fundamental surprises with broad significance, including the findings that: 1) Natural use of an unconventional gene regulatory mechanism based on timed toggling between two transcript isoforms of different translatability is common in meiosis and beyond (Cheng and Otto et al. Cell, 2018; Van Dalfsen et al. Developmental Cell 2018; Hollerer et al. G3 2019); 2)Protein degradation is a key driver of meiotic protein levels and, as a rule, protein complex members are synthesized imprecisely and matched by protein degradation during meiosis (Eisenberg and Higdon et al. Cell Reports, 2018); 3) Over 2000 novel short proteins are synthesized in a regulated manner during meiosis (Brar et al. Science, 2012; Ingolia et al. Cell Reports, 2014; Hollerer et al. Proteomics, 2018) 4) Yeast cells synthesize hundreds of alternative protein isoforms (Brar et al. Science, 2012; Eisenberg et al. Cell Systems 2020); and 5) Changes in the expression of the protein components of the ribosome are linked to characteristic gene expression signatures in meiosis and beyond (Cheng et al. Molecular Cell 2019; Cheng et al., Nucleic Acids Research 2019).

A global view of meiotic protein synthesis. Ribosome footprints over each S. cerevisiae gene are shown as columns. Each time point is shown as a row, with 25 meiotic timepoints shown and two mitotic (exponential) controls. Cartoons on the left show progression through meiosis. Note the variety of patterns of regulation of protein synthesis through meiosis, with nearly every gene in the yeast genome expressed and highly regulated. This regulation is mediated by both transcriptional and hundreds of newly identified examples of dramatic translational control. At the right are more detailed views of two genes, with both mRNA and ribosome footprints shown on pooled genome browser tracks. Note that SPS1 and SPS2 show similar patterns of mRNA abundance, but very different translation patterns, reflecting just one of these examples of strong translational control.
Ongoing projects in the Brar lab
These discoveries have motivated several research directions in our lab that are focused on answering fundamental questions about gene regulation through meiosis.
Some questions that interest us are:
Read more about any of these projects by clicking above or on the icons at the top of the page.